Gene-centric multiomics clustering
Thomas Rauter
19 January, 2026
Source:vignettes/Gene_centric_multiomics_clustering.Rmd
Gene_centric_multiomics_clustering.RmdGene-centric multiomics clustering
This vignette demonstrates the gene-centric multi-omics clustering
approach implemented in
SplineOmics::cluster_genes_multiomics() and shows how to
visualize the results using
SplineOmics::plot_umap_clusters().
Figure 1. Overview of the gene-centric multi-omics
clustering approach.
See the figure here.
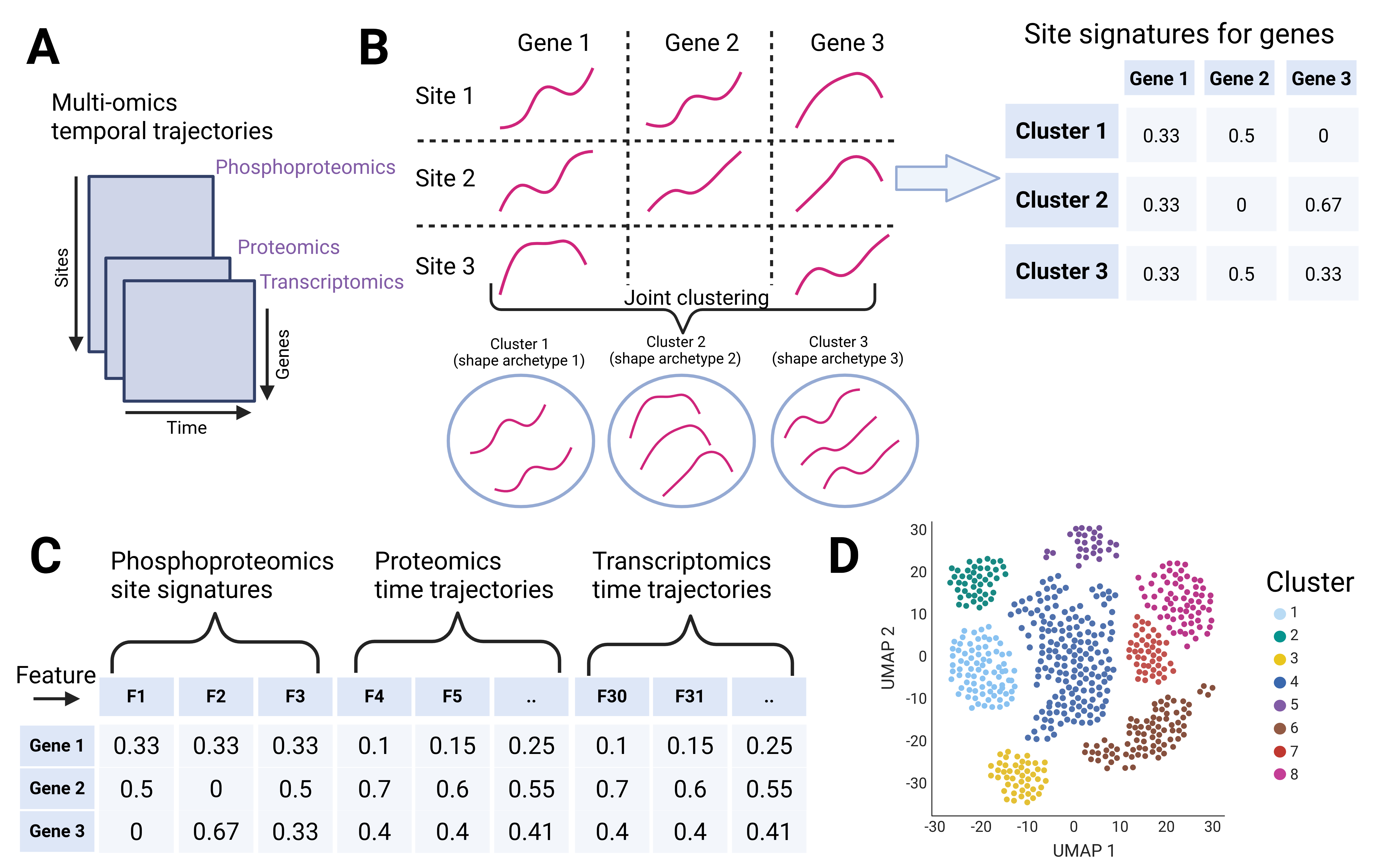{kind=link}
The example below uses a small synthetic dataset with two experimental conditions and three molecular modalities, including a many-to-one feature-level modality.
1. Simulate multi-omics time-series data
We simulate expression trajectories for 40 genes measured at 9 time points. Each gene is assigned to one of five latent temporal patterns used only for data generation.
set.seed(1)
n_genes <- 40
genes <- paste0("gene", seq_len(n_genes))
n_tp <- 9
t <- seq(0, 1, length.out = n_tp)
.add_noise <- function(x, sd = 0.15) {
x + rnorm(length(x), mean = 0, sd = sd)
}
shapes <- list(
sin = sin(2 * pi * t),
cos = cos(2 * pi * t),
up = 2 * t - 1,
down = 1 - 2 * t,
peak = exp(-((t - 0.5) / 0.18)^2) * 2 - 1
)
true_k <- 5
gene_group <- sample(seq_len(true_k), n_genes, replace = TRUE)1.1 Simulate RNA and protein (one-to-one modalities)
RNA is generated directly from the latent shapes. Protein trajectories are simulated as lagged and scaled versions of RNA, mimicking delayed protein dynamics.
.simulate_one_to_one <- function(
genes,
shapes,
gene_group,
noise_sd = 0.18
) {
n_genes <- length(genes)
n_tp <- length(shapes[[1]])
X <- matrix(
NA_real_,
nrow = n_genes,
ncol = n_tp,
dimnames = list(genes, paste0("tp", seq_len(n_tp)))
)
for (i in seq_len(n_genes)) {
k <- gene_group[i]
X[i, ] <- .add_noise(shapes[[k]], sd = noise_sd)
}
X
}
rna_ctrl <- .simulate_one_to_one(
genes = genes,
shapes = shapes,
gene_group = gene_group,
noise_sd = 0.16
)
rna_treat <- rna_ctrl
for (i in seq_len(n_genes)) {
k <- gene_group[i]
rna_treat[i, ] <-
.add_noise(shapes[[k]] + 0.15 * t, sd = 0.16)
}
protein_ctrl <- rna_ctrl
protein_treat <- rna_treat1.2 Simulate phospho features (many-to-one modality)
For each gene, multiple phospho features are generated. Rows are
named <gene>_<feature> to encode the
many-to-one mapping.
.simulate_many_to_one <- function(
genes,
shapes,
gene_group,
min_feat = 1,
max_feat = 4,
noise_sd = 0.20
) {
feat_counts <- sample(
min_feat:max_feat,
length(genes),
replace = TRUE
)
feat_ids <- unlist(
mapply(
function(g, n) paste0(g, "_p", seq_len(n)),
genes,
feat_counts,
SIMPLIFY = FALSE
),
use.names = FALSE
)
n_tp <- length(shapes[[1]])
X <- matrix(
NA_real_,
nrow = length(feat_ids),
ncol = n_tp,
dimnames = list(feat_ids, paste0("tp", seq_len(n_tp)))
)
idx <- 0L
for (i in seq_along(genes)) {
base <- shapes[[gene_group[i]]]
for (j in seq_len(feat_counts[i])) {
idx <- idx + 1L
X[idx, ] <- .add_noise(base, sd = noise_sd)
}
}
X
}
phospho_ctrl <- .simulate_many_to_one(
genes,
shapes,
gene_group
)
phospho_treat <- phospho_ctrl2. Assemble inputs for clustering
Each condition is represented as a list of modality matrices. The
meta data frame describes modality properties shared across
conditions.
data <- list(
Ctrl = list(
rna = rna_ctrl,
protein = protein_ctrl,
phospho = phospho_ctrl
),
Treat = list(
rna = rna_treat,
protein = protein_treat,
phospho = phospho_treat
)
)
meta <- data.frame(
modality = c("rna", "protein", "phospho"),
many_to_one_k = c(NA_real_, NA_real_, 4),
modality_w = c(1, 1, 1),
stringsAsFactors = FALSE
)3. Run gene-centric multi-omics clustering
Clustering is performed on the UMAP neighborhood graph derived from the gene-centric representation.
res <- cluster_genes_multiomics(
data = data,
meta = meta,
k = 5L,
gene_mode = "intersection",
n_neighbors = 15L,
verbose = TRUE
)## [cluster_genes_multiomics] total runtime:
## • 1.2 secs
cluster_table <- res$cluster_table4. Visualize clusters in UMAP space
The UMAP embedding returned by the clustering function can be
visualized using plot_umap_clusters().
p <- plot_umap_clusters(
cluster_table = cluster_table,
umap_embedding = res$umap_fit$embedding,
point_size = 1.2
)
print(p)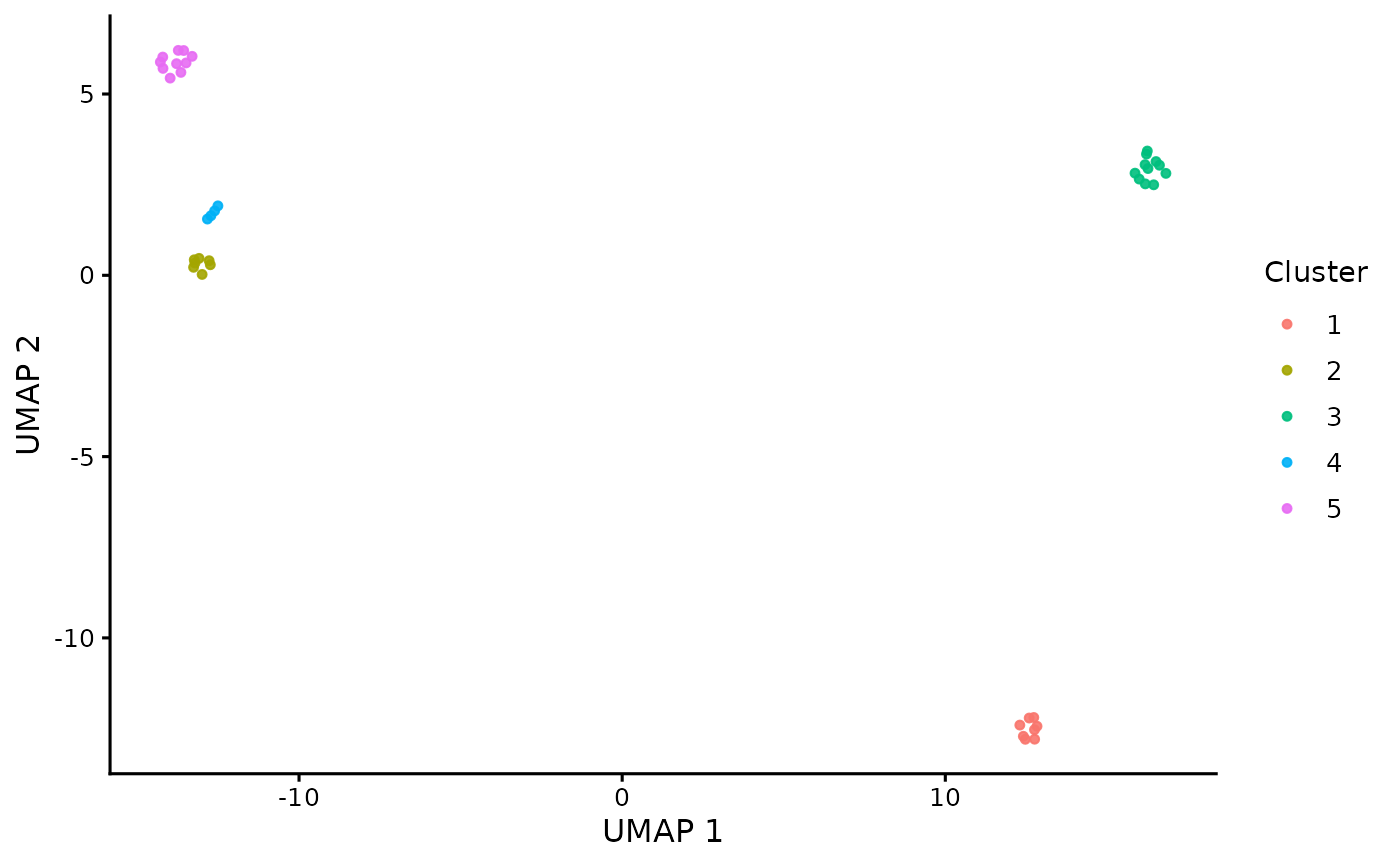
Session information
## R version 4.5.3 (2026-03-11)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 22.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0 LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=de_AT.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=de_AT.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=de_AT.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=de_AT.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Europe/Vienna
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices datasets utils methods base
##
## other attached packages:
## [1] ggplot2_4.0.2 tibble_3.3.1 SplineOmics_0.4.4
##
## loaded via a namespace (and not attached):
## [1] Rdpack_2.6.5 bitops_1.0-9 pbapply_1.7-4
## [4] writexl_1.5.4 rlang_1.1.7 magrittr_2.0.4
## [7] clue_0.3-66 GetoptLong_1.1.0 RcppAnnoy_0.0.23
## [10] otel_0.2.0 matrixStats_1.5.0 compiler_4.5.3
## [13] reshape2_1.4.5 png_0.1-8 systemfonts_1.3.1
## [16] vctrs_0.7.1 stringr_1.6.0 pkgconfig_2.0.3
## [19] shape_1.4.6.1 crayon_1.5.3 fastmap_1.2.0
## [22] backports_1.5.0 labeling_0.4.3 caTools_1.18.3
## [25] rmarkdown_2.30 nloptr_2.2.1 ragg_1.5.0
## [28] purrr_1.2.1 xfun_0.56 cachem_1.1.0
## [31] jsonlite_2.0.0 progress_1.2.3 EnvStats_3.1.0
## [34] remaCor_0.0.20 gmp_0.7-5 BiocParallel_1.42.2
## [37] broom_1.0.12 parallel_4.5.3 prettyunits_1.2.0
## [40] cluster_2.1.8.2 R6_2.6.1 stringi_1.8.7
## [43] bslib_0.10.0 RColorBrewer_1.1-3 limma_3.64.3
## [46] boot_1.3-32 car_3.1-5 ClusterR_1.3.6
## [49] numDeriv_2016.8-1.1 jquerylib_0.1.4 Rcpp_1.1.1
## [52] iterators_1.0.14 knitr_1.51 base64enc_0.1-6
## [55] IRanges_2.42.0 Matrix_1.7-4 splines_4.5.3
## [58] tidyselect_1.2.1 rstudioapi_0.18.0 abind_1.4-8
## [61] yaml_2.3.12 doParallel_1.0.17 gplots_3.3.0
## [64] codetools_0.2-19 plyr_1.8.9 lmerTest_3.2-0
## [67] lattice_0.22-9 withr_3.0.2 Biobase_2.68.0
## [70] S7_0.2.1 evaluate_1.0.5 desc_1.4.3
## [73] zip_2.3.3 circlize_0.4.17 pillar_1.11.1
## [76] BiocManager_1.30.27 carData_3.0-6 KernSmooth_2.23-26
## [79] checkmate_2.3.4 renv_1.1.7 foreach_1.5.2
## [82] stats4_4.5.3 reformulas_0.4.4 generics_0.1.4
## [85] S4Vectors_0.46.0 hms_1.1.4 scales_1.4.0
## [88] aod_1.3.3 minqa_1.2.8 gtools_3.9.5
## [91] RhpcBLASctl_0.23-42 glue_1.8.0 tools_4.5.3
## [94] fANCOVA_0.6-1 variancePartition_1.38.1 RSpectra_0.16-2
## [97] lme4_1.1-38 mvtnorm_1.3-3 fs_1.6.6
## [100] grid_4.5.3 tidyr_1.3.2 rbibutils_2.4.1
## [103] colorspace_2.1-2 nlme_3.1-168 Formula_1.2-5
## [106] cli_3.6.5 textshaping_1.0.4 svglite_2.2.2
## [109] ComplexHeatmap_2.24.1 dplyr_1.2.0 uwot_0.2.4
## [112] corpcor_1.6.10 gtable_0.3.6 sass_0.4.10
## [115] digest_0.6.39 BiocGenerics_0.54.1 pbkrtest_0.5.5
## [118] ggrepel_0.9.6 rjson_0.2.23 htmlwidgets_1.6.4
## [121] farver_2.1.2 htmltools_0.5.9 pkgdown_2.2.0
## [124] lifecycle_1.0.5 GlobalOptions_0.1.3 statmod_1.5.1
## [127] MASS_7.3-65